
Overview
EstiAge is a simple likelihood based method to estimate the age of the most recent common ancestor of individuals, based on the information provided by their shared haplotypes.
EstiAge can be used on Microsatellite data as well as NGS data.
How it works
When estimating the age of a genetic variant, EstiAge assumes that all affected samples descended from a common ancestor who introduced the variant n generations ago. An estimate of n is obtained from the size of the haplotype shared by individuals on both sides of the variant locus by finding the most likely positions of recombinations on the ancestral haplotype in the different samples. Then, the value of n is converted to age in years by multiplying by the assumed 25 years per generation.
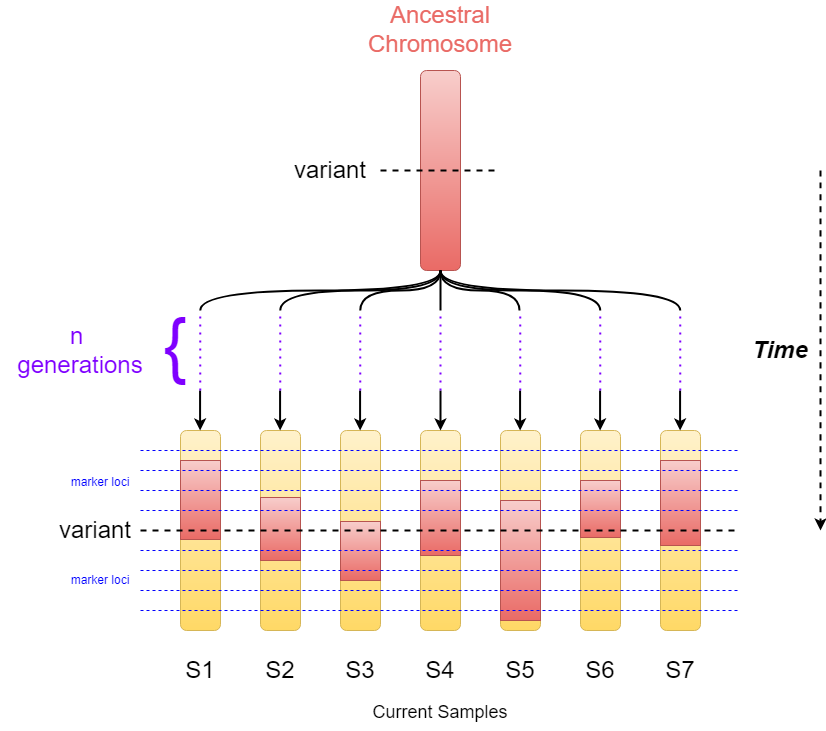
This method is based on LD decay and rely on the fact that the variant will be in LD with polymorphic marker alleles. Recombination will decay this LD over time and can be used to date the variant. This approach relies on the fact that individuals carrying a variant have inherited it as segments of the ancestral chromosome that originally acquired the variants. These segments will have different size due to recombination events occurring randomly. The size of the segments decreasing over time, their conserved lengths can be used to estimate the variant's age (short segments will imply older variants).
Initially, Microsatellite data were used as Markers. A new implementation now allows the user to estimate the age of a variant from a phased VCF File. In this case, homozygous variant alleles will be used as markers.